
Tutorial
Tutorial
We will demonstrate an example run of PhaBOX2 on a small dataset here.
The example output can be found via EXAMPLE.
🪜 Prepare a submission
✅ STEP 1 - Enter your input sequences
First, let's download the example dataset exmaple_contigs.fa, containing 390 sequences:
Clik on [Download the example file] on the webpage Or wget https://github.com/KennthShang/PhaBOX/releases/download/v2.1.0/example_contigs.fa
A screenshot of the exmaple_contigs.fa should look like:
>example_0 AGATACTAACTCTGCTGCATAGACAAGAAATTCGTCTTTGCGGGAATATTTACCTGCAAG GTATATTTTCACATTAACCTCTCAAAAAGCGTTTAACCACTGCTGGTACAACCCCATTTC ·······
Please select the FASTA file that you wish to upload. Note: If you have the FASTA file of predicted proteins (generated by prodigal), you can upload it to speed up PhaBOX.
✅ STEP 2 - Select your program
Comprehensive end-to-end pipeline
The next step is to run phabox2 to classify these contigs. There are nine programs for users.
Select a program to run:
end_to_end || Run phamer, phagcn, phatyp, and cherry once (default)
phamer || Virus identification
phagcn || Taxonomy classification
phatyp || Lifestyle prediction
cherry || Host prediction
contamination || Contamination/proviurs detection
votu || vOTU grouping (ANI-based or AAI-based)
tree || Build phylogenetic trees based on marker genes
✅ STEP 3 - Set your parameters
Here, we demonstrate the basic usage of running phabox2 in end_to_end mode.
There are several options used in end_to_end mode:
--len
Filter the length of contigs || default: 3000
Contigs with length smaller than this value will not proceed
--reject
Reject sequences in which the percent proteins aligned to known phages is smaller than the value.
Default: 10
Range from 0 to 20
--aai
Average amino acids identity || default: 75 || range from 0 to 100
--share
Minimum shared number of proteins || default: 15 || range from 0 to 100
--pcov
Protein-based coverage || default: 80 || range from 0 to 100
--ccov
Alignment identity for CRISPRs || default: 90 || range from 90 to 100
--ccoverage
Alignment coverage for CRISPRs || default: 90 || range from 0 to 100
All the default parameters are optimized based on the latest ICTV 2024. But you can adjust them according to your needs.
Detailed options for different programs can be found in the Parameter Options. We also provide hints for you when adjusting the parameters. You can easily access the explanations of the parameters by touching the icon after the parameters.
🚀 Ready to go!
 |
Please select whether you wish to be notified by email. If yes, turn on the button and paste your email address. Otherwise, submit your task directly and remember the job ID.
Once you submit your job, the webpage will jump to the Result page. Your job ID of your submission and a process bar will be shown on the page. Please remember your job ID to find your results.
Sometimes you will find your work is stuck with a notification The task is pending. This is because other users' tasks are running. It is estimated that a 350Mb file might take 30 minutes to run. Please be patient. We also show the queue of the running/pending tasks on the page. To protect the privacy, we masked some digits of the job ID, but you can search your job using your IDs. If you want to run PhaBOX on your large-scale data, we suggest you to turn on the email notification OR you can try our local version which allows more threads to speed up the program.
📈 Visualize and download your results!
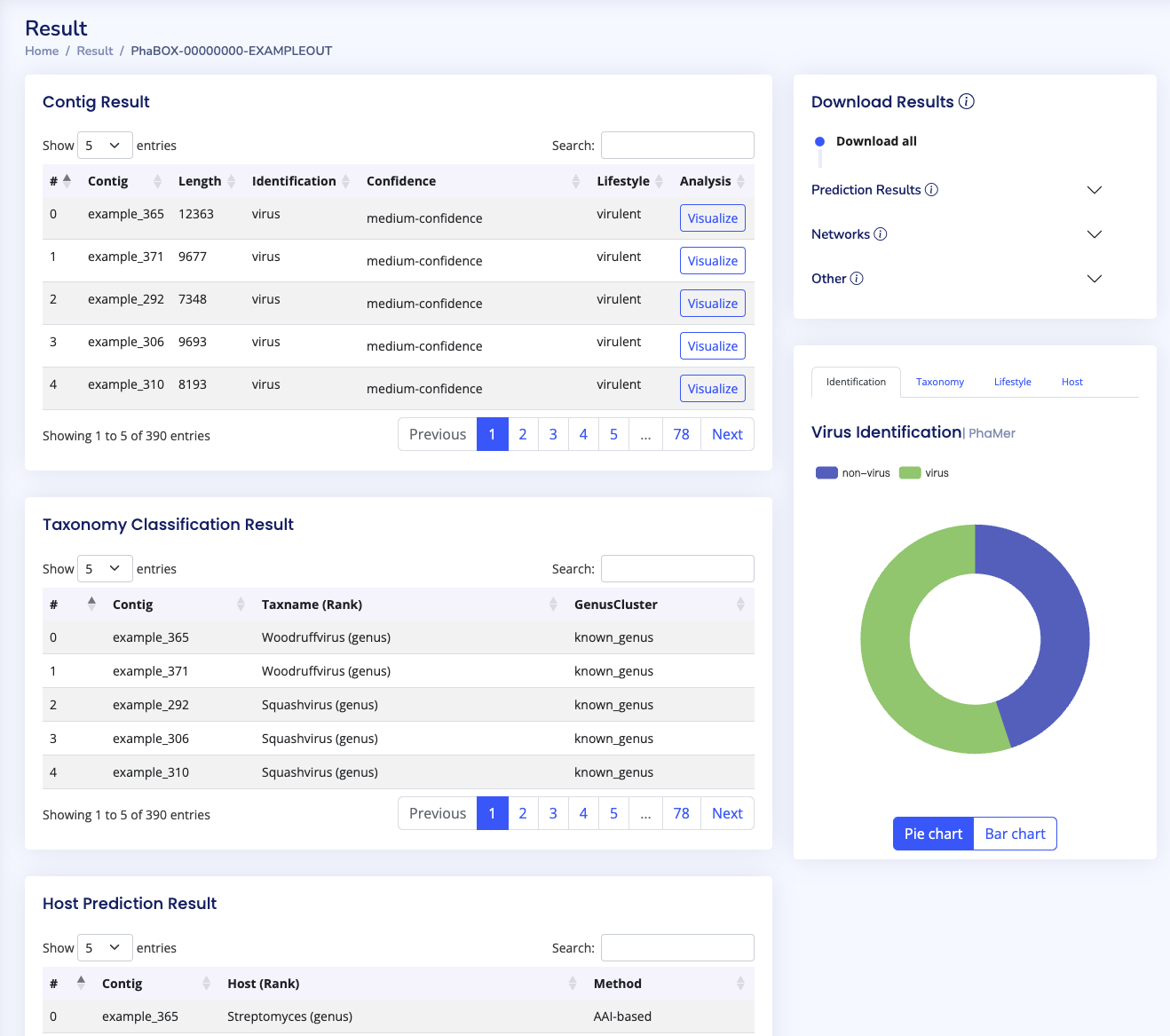 |
Once your program is finished, you can access your results via the search bar. Again, an example can be found via EXAMPLE. If you want to visualize your results, please click on the visualize button.
In the end_to_end mode, there are four main visualization blocks showing the results of virus identification, lifestyle prediction, taxonomy classification, and host prediction. Please note that prediction with non-virus and low-confidence will not be used in the following taxonomy, host, and lifestyle prediction tasks. Alternatively, we provide a file named uncertain_sequences_for_contamination_task.fa for you to run the contamination task and check the quality of these sequences
A further visualization can be accessed via the visualize buttons in the Contig Result block. This will show the protein annotations for your chosen sequence. You can also touch the proteins to review detailed information.
Except for the visualization, you can download all the results within the Download Results bar. Detailed explanation of the format can be found in the Outputs Formats.